ANSM - Mis à jour le : 10/12/2007
Concerne les médicaments pouvant être obtenus uniquement sur ordonnance :
Veuillez lire attentivement l'intégralité de cette notice avant d’utiliser ce médicament.
Gardez cette notice, vous pourriez avoir besoin de la relire.
Si vous avez d’autres questions, si vous avez un doute, demandez plus d’informations à votre médecin ou à votre pharmacien.
Ce médicament vous a été personnellement prescrit. Ne le donnez jamais à quelqu’un d’autre, même en cas de symptômes identiques, car cela pourrait lui être nocif.
Dénomination du médicament
ENDOBULINE 50 mg/ml, poudre et solvant pour solution injectable
Immunoglobuline humaine normale
Liste complète des substances actives et des excipients
Dans 1 ml de solution reconstituée :
La substance active est :
Immunoglobuline humaine normale (IV)................................................................................................. 50 mg*
* correspondant à une quantité totale de protéines, dont au moins 95% sont des IgG.
Après reconstitution
·Un flacon de 200 ml contient 10 g d’immunoglobuline humaine normale
|
Répartition en sous-classes d'IgG : |
|
|
IgG1 |
50 - 80 % |
|
IgG2 |
20 - 50 % |
|
IgG3 |
0 % |
|
IgG4 |
1 - 3 % |
|
IgA |
max. 1mg/g de protéines |
|
Trypsine d'origine animale |
traces |
Les autres composants sont :
Poudre : glucose, chlorure de sodium.
Solvant : eau pour préparations injectables.
Nom et adresse du titulaire de l'autorisation de mise sur le marché et du titulaire de l'autorisation de fabrication responsable de la libération des lots, si différent
Titulaire/ Exploitant
LABORATOIRE BAXTER S.A.S
6, avenue Louis Pasteur
B.P. 56
78311 MAUREPAS Cedex
Fabricant
BAXTER A.G.
Industriestrasse 67
A-1221 VIENNE
AUTRICHE
Forme pharmaceutique et contenu ; classe pharmacothérapeutique
Endobuline se présente sous la forme d'une poudre et solvant pour solution injectable (flacon de 10 ml, 20 ml, 50 ml, 100 ml et 200 ml).
Poudre et solvant pour solution injectable (flacon de 10 ml, 20 ml, 50 ml, 100 ml et 200 ml) :
Ce médicament est une immunoglobuline humaine normale.
Il est indiqué dans :
le traitement de substitution :
le traitement immunomodulateur :
2. Quelles sont les informations nécessaires avant d’utiliser ENDOBULINE 50 mg/ml, poudre et solvant pour solution injectable ?
Liste des informations nécessaires avant la prise du médicament
Sans objet.
Contre-indications
Ne pas utiliser ENDOBULINE :·si vous avez déjà présenté une réponse allergique connue à l'un des constituants de la préparation.
Précautions d'emploi ; mises en garde spéciales
Vous devez utiliser ENDOBULINE avec précaution :
·en cas d'insuffisance rénale (altération de la fonction du rein), si vous avez plus de 65 ans ou si vous avez une surcharge pondérale.
·en cas de diabète latent, diabète ou régime hypoglucidique (pauvre en sucres).
En cas de doute, ne pas hésiter à demander l'avis de votre médecin ou de votre pharmacien.
Certains effets indésirables peuvent être associés au débit d’administration. Le débit recommandé (voir paragraphe Mode d'administration) doit être scrupuleusement observé et les patients doivent rester sous surveillance pendant toute la durée de la perfusion afin de détecter d'éventuels signes d'intolérance.
Le risque de réactions allergiques généralisées, voire d'état de choc, est plus fréquent :
·en cas de perfusion intraveineuse rapide (voir paragraphe Mode d'administration),
·chez les patients hypo- ou agammaglobulinémiques (avec défaut de production d'anticorps), avec ou sans déficit en IgA, et plus particulièrement lors de la première perfusion d'ENDOBULINE, ou lorsque le dernier traitement par ENDOBULINE remonte à plus de 8 semaines.
Les vraies réponses allergiques à ce médicament sont rares. Une intolérance aux immunoglobulines peut se développer dans les très rares cas de déficit en IgA où le patient possède des anticorps anti-IgA.
Très rarement, ENDOBULINE peut entraîner une chute brutale de la pression artérielle associée à une allergie généralisée même chez les patients qui ont présenté une bonne tolérance à une administration précédente d'immunoglobuline humaine normale.
Les complications potentielles peuvent être souvent évitées ; il est souhaitable :
·de surveiller attentivement le débit des perfusions ;
·de s'assurer initialement de la tolérance de l'administration d'ENDOBULINE par une perfusion lente (0,5 ml/kg/h) ;
·chez les patients ayant déjà reçu des immunoglobulines humaines normales, le débit sera adapté en fonction de la tolérance clinique, sans dépasser un débit de 0,5 ml/kg/h pendant la première demi-heure, puis en augmentant progressivement sans dépasser 4 ml/kg/h ;
·de tenir compte de la teneur en sucre (1 g/g d'ENDOBULINE) en cas de diabète latent, de diabète ou de régime hypoglucidique (régime pauvre en sucres) ;
·de garder les patients sous surveillance pendant toute la durée de la perfusion, afin de détecter d'éventuels signes d'intolérance.
Des cas d'insuffisance rénale aiguë (altération du fonctionnement du rein) ont été rapportés chez des patients recevant de l'immunoglobuline normale. Dans la plupart des cas, des facteurs de risque ont été identifiés, tels une insuffisance rénale pré-existante, un diabète, une hypovolémie ou une obésité, la prise concomitante de médicaments néphrotoxiques ou un âge supérieur à 65 ans.
Chez ces patients, l'administration d'ENDOBULINE impose :
·une hydratation correcte avant l'administration d'ENDOBULINE,
·de surveiller la diurèse (volume d'urine émis par 24 heures),
·de doser la créatininémie (mesure du fonctionnement du rein),
·d'éviter d'associer certains diurétiques.
Bien que ces cas d'insuffisance rénale aient été associés à l'utilisation de nombreuses spécialités d'immunoglobuline normale, celles contenant du saccharose comme stabilisant représentent la plus large part.
Aussi, chez les patients à risque, l'utilisation de préparations d'immunoglobuline normale ne contenant pas de saccharose, comme ENDOBULINE, doit être envisagée.
Ce médicament contient 24 mg de sodium par gramme d'immunoglobuline : en tenir compte chez les personnes suivant un régime pauvre en sel.
En cas de réactions de type allergique ou allergique généralisée, il convient d'interrompre immédiatement la perfusion. En cas de choc, le traitement symptomatique relatif à l'état de choc devra être instauré.
Le patient doit être maintenu en observation pendant au moins 20 minutes après la fin de la perfusion. En cas de première perfusion d'ENDOBULINE, le patient doit être maintenu en observation pendant au moins 1 heure après la fin de la perfusion.
Lorsque les médicaments sont préparés à partir de sang ou de plasma humain sotn administrés, le risque de transmission de maladies infectieuses ne peut-être totalement exclu.
Ceci s'applique également à tous les virus inconnus ou émergents ou d’autres types d’agents infectieux.
Les mesures prises sont considérées comme efficaces pour lutter contre le risque d’infection par le virus de l’immunodéficience humaine (VIH), le virus de l’hépatite B, le virus de l’hépatite C, le virus de l’hépatite A et le parvovirus B19.
Les immoglobulines humaines normales ne sont pas associées aux infections par le virus de l’hépatite A et le parvovirus B19, probablement grâce aux anticorps protecteurs présents dans ce produit.
Interactions avec les aliments et les boissons
Sans objet.
Utilisation pendant la grossesse et l'allaitement
GrossesseAucune étude de reproduction chez l'animal n'a été conduite avec ENDOBULINE et l'expérience chez la femme enceinte est limitée. Bien qu'aucune réaction indésirable sur le fœtus n'ait été observée, ENDOBULINE ne doit être administrée qu'en cas de nécessité bien établie.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
AllaitementLes protéines contenues dans ENDOBULINE étant des constituants normaux du plasma humain, leur passage dans le lait maternel ne doit pas provoquer d'effets indésirables chez le nouveau‑né.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Sans objet.
Effets sur l'aptitude à conduire des véhicules ou à utiliser des machines
Conduite de véhicules et utilisation de machines :Rien ne suggère qu'ENDOBULINE diminue l'aptitude à conduire des véhicules et à utiliser des machines.
Liste des excipients à effet notoire
Liste des excipients à effet notoire : glucose, sodium.
Interaction avec d'autres médicaments
Prise ou utilisation d'autres médicaments :Vaccins constitués de virus vivants atténués
L'administration d'immunoglobuline humaine normale peut entraver l'efficacité des vaccins constitués de virus vivants atténués tels que les vaccins contre la rougeole, la rubéole, les oreillons et la varicelle. Après perfusion de ce médicament, attendre au minimum 6 semaines (de préférence 3 mois) avant d'administrer ce type de vaccins.
Si le patient a reçu des vaccins constitués de virus vivants atténués (rougeole, rubéole, oreillons, varicelle) au cours des 2 semaines précédant la perfusion, un contrôle des anticorps protecteurs post-vaccinaux peut être nécessaire en vue d'un éventuel rappel.
Interférence avec des tests sérologiques
Après administration d'immunoglobuline humaine normale, l'augmentation transitoire de la concentration de divers anticorps transférés peut être responsable de sérologies positives temporaires.
Ce médicament contenant des anticorps anti-érythrocytaires (anticorps contre les globules rouges), son administration peut être suivie de façon transitoire d'un test de Coombs positif (test effectué pour détecter la présence de ces anticorps).
Veuillez indiquer à votre médecin ou à votre pharmacien si vous prenez ou avez pris récemment un autre médicament, même s'il s'agit d'un médicament obtenu sans ordonnance.
3. COMMENT UTILISER ENDOBULINE 50 mg/ml, poudre et solvant pour solution injectable?
Instructions pour un bon usage
Sans objet.
Posologie, Fréquence d'administration et Durée du traitement, Mode et/ou voie(s) d'administration
Posologie
Si vous avez l'impression que l'effet d'ENDOBULINE est trop fort ou trop faible, consultez votre médecin ou votre pharmacien.
La posologie et l'intervalle entre les administrations dépendent de l'usage auquel est destiné le traitement (substitution) et de la demi-vie de l'Immunoglobuline humaine normale par voie intraveineuse (IgIV) in vivo chez les patients atteints de déficit immunitaire.
Les posologies suivantes sont données à titre indicatif.
Traitement de substitution en cas de déficit immunitaire primitif :Le traitement doit avoir pour but d'assurer un taux d'IgG résiduel (c'est-à-dire avant l'administration suivante d'immunoglobuline G humaine normale) d'au moins 4 à 6 g/l. Après le début d'un traitement par l'immunoglobuline humaine normale, l'équilibre s'effectue en 3 à 6 mois. On peut recommander une dose de charge de 0,4 à 0,8 g/kg selon les circonstances (infection) puis une perfusion de 0,2 g/kg toutes les 3 semaines. Les doses d'immunoglobuline humaine normale nécessaires pour atteindre un taux résiduel de 4 à 6 g/l sont de l'ordre de 0,3 g/kg/mois, avec des extrêmes de 0,2 à 0,8 g/kg/mois. L'intervalle des perfusions varie de 15 jours à 1 mois. La survenue d'infections peut nécessiter l'emploi temporaire de perfusions plus fréquentes.
Dans le traitement substitutif des déficits immunitaires primitifs, un dosage des concentrations sériques d'IgG avant chaque perfusion s'avère nécessaire pour contrôler l'activité du traitement et éventuellement ajuster la dose ou l'intervalle d'administration.
Traitement de substitution en cas de déficit immunitaire secondaire :On peut recommander une dose de 0,2 à 0,4 g/kg toutes les 3 à 4 semaines.
Purpura thrombopénique idiopathique (PTI) :Pour le traitement d'attaque, 0,8 à 1g/kg/j au jour 1, éventuellement répété au jour 3, ou 0,4g/kg/j pendant 2 à 5 jours. Ce traitement peut être renouvelé en cas de réapparition d'une thrombopénie sévère.
Syndrome de Guillain Barré de l'adulte :0,4g/kg de poids corporel/jour pendant 5 jours.
Maladie de Kawasaki :1,6 à 2,0 g/kg administrés en plusieurs doses réparties sur 2 à 5 jours ou 2,0 g/kg en dose unique, associées à l'acide acétylsalicylique.
Allogreffe de cellules souches hématopoïétiques :Les IgIV sont généralement utilisées avant et après l'allogreffe.
Les IgIV ont un effet préventif vis-à-vis de la survenue de complications infectieuses et réduisent la fréquence et la sévérité de la maladie du greffon contre l'hôte chez les receveurs d'allogreffe de cellules souches hématopoïétiques.
La posologie est déterminée sur la base individuelle et commence habituellement par une dose de 0,5 g/kg/semaine de J‑7 à J+90.
En cas de défaut persistant de la production d'anticorps, on recommande actuellement la posologie de 0,5 g/kg/mois jusqu'à J+360.
Posologie et Mode d'administration| Indication | Posologie | Rythme des injections | Traitement associé |
|
Traitement substitutif dans les déficits immunitaires primitifs |
- dose de charge : 0,4 à 0,8 g/kg |
||
|
- dose d'entretien : 0,2 à 0,8 g/kg |
toutes les 2 à 4 semaines pour obtenir un taux résiduel d'IgG d'au moins 4 à 6 g/l |
||
|
Traitement substitutif dans les déficits immunitaires secondaires |
0,2 à 0,4 g/kg |
toutes les 3 à 4 semaines pour obtenir un taux résiduel d'IgG d'au moins 4 à 6 g/l |
|
|
Allogreffes de cellules souches hématopoïétiques. |
|||
|
Prévention des complications infectieuses et de la maladie du greffon contre l'hôte |
0,5 g/kg |
chaque semaine de J-7 à J + 90 |
|
|
En cas de défaut persistant de la production d'anticorps |
0,5 g/kg |
chaque mois jusqu'à J + 260 |
|
|
Traitement immunodulateur |
|||
|
Purpura thrombopénique idiopathique |
0,8 à 1g/kg |
A J1, éventuellement |
|
|
ou |
répété à J3 |
||
|
0,4 g/kg/j |
pendant 2 à 5 jours |
||
|
Syndrome de Guillain et Barré de l'adulte |
0,4 g/kg/j |
pendant 5 jours |
|
|
Maladie de Kawasaki |
1,6 à 2,0 g/kg |
en plusieurs doses |
acide acétylsalicylique |
|
ou |
réparties sur 2 à 5 j |
||
|
2g/kg |
en une dose unique |
ENDOBULINE se présente sous la forme d'une poudre à reconstituer extemporanément avec de l'eau pour préparations injectables.
Reconstitution : utiliser une technique aseptiqueNe jamais utiliser les flacons dès la sortie du réfrigérateur. Amener le flacon de solvant (eau pour préparations injectables) à température ambiante.
La dissolution complète doit être obtenue en moins de 10 minutes. Une agitation douce à une température de 37°C maximum permet de réduire le temps nécessaire à la dissolution.
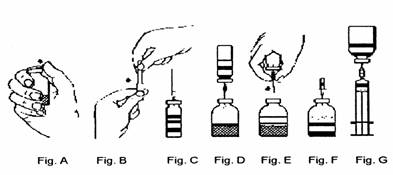
Présentations de 0,5 g et 1 g :
1. Retirer la capsule de protection des flacons de solvant et de poudre (fig. A) et désinfecter les bouchons de chaque flacon.
2. Retirer le capuchon protecteur d’une des extrémités du dispositif de transfert fourni et introduire l’extrémité libre dans le bouchon du flacon de solvant (fig. B et fig. C).
3. Retirer le capuchon protecteur de l’autre extrémité du dispositif de transfert. Ne pas toucher l’extrémité libre.
4. Retourner l’ensemble flacon de solvant/dispositif de transfert en le plaçant au-dessus du flacon de poudre et introduire verticalement l’extrémité libre du dispositif de transfert au centre du bouchon du flacon de poudre (fig. D). Par une légère rotation, ajuster le système de transfert sur le flacon de poudre. Le solvant est aspiré dans le flacon de poudre par le vide.
5. Séparer les deux flacons en retirant l’ensemble flacon de solvant/dispositif de transfert du flacon de poudre (fig. E). Agiter modérément le flacon de poudre avec un mouvement de rotation doux pour accélérer la dissolution. Ne pas secouer le flacon.
6. Après reconstitution de la poudre lyophilisée, introduire l’aiguille d’aération fournie (fig. F) pour éliminer la mousse qui se serait formée. Retirer l’aiguille d’aération.
|
|
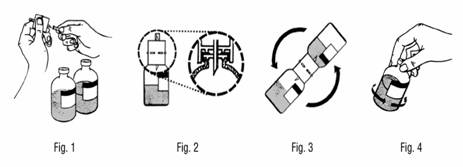
Présentations de 2,5 g, 5 g et 10 g :
1. Retirer la capsule de protection des flacons de solvant et de poudre et désinfecter les bouchons de chaque flacon.
2. Retirer le capuchon protecteur d’une des extrémités du dispositif de transfert fourni (fig. 1) et introduire verticalement l’extrémité libre au centre du bouchon du flacon de solvant. Appuyer fermement afin que le dispositif de transfert s’emboîte parfaitement dans le flacon de solvant. (fig. 2).
Attention : si l’insertion ne se fait pas au centre du bouchon, celui-ci est susceptible de se déloger.
3. Retirer le capuchon protecteur de l’autre extrémité du dispositif de transfert. Ne pas toucher l’extrémité libre.
4. Maintenir le flacon de poudre fermement et à un angle d’environ 45 degrés. Retourner l’ensemble flacon de solvant/dispositif de transfert dans l’axe du flacon de poudre et introduire fermement le dispositif de transfert dans le flacon de poudre au centre du bouchon (fig. 3). Le solvant est aspiré dans le flacon de poudre par le vide.
Note : retourner l’ensemble flacon de solvant/dispositif de transfert et l’introduire dans le flacon de poudre rapidement afin d’éviter la perte de solvant.
Attention : si l’insertion ne se fait pas au centre du bouchon, celui-ci est susceptible de se déloger et une perte de vide peut se produire.
5. Séparer les deux flacons en retirant l’ensemble flacon de solvant/dispositif de transfert du flacon de poudre. Agiter modérément le flacon de poudre avec un mouvement de rotation doux pour accélérer la dissolution (fig. 4). Ne pas secouer le flacon.
|
|
Le produit reconstitué doit être examiné à l'œil, afin de s'assurer qu'il ne contient pas de particules et qu'il n'est pas décoloré avant administration.
Ne pas utiliser de solutions présentant un aspect non homogène ou contenant un dépôt.
Présentation de 0,5 g et 1 g :
Présentation de 2,5 g, 5 g et 10 g :
Dans le cas où un dispositif de perfusion différent de celui fourni avec ENDOBULINE 50 mg/ml est utilisé, il faut s’assurer que ce dispositif est muni d’un filtre adéquat pour prévenir le risque d’administration de fines particules issues du bouchon (risque de micro embole).
ENDOBULINE peut être administrée en même temps qu’une solution salée isotonique (Solution de chlorure de sodium à 0,9 pour cent). Si une concentration plus faible d’Endobuline 50 mg/ml est désirée, une dilution peut être effectuée avec une solution de chlorure de sodium isotonique (NaCl 0,9%).
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Symptômes et instructions en cas de surdosage
Si vous avec utilisé plus d'ENDOBULINE 50 mg/ml, poudre et solvant pour solution injectable que vous n’auriez dû : consultez immédiatement votre médecin ou votre pharmacien.
Si aucun effet indésirable spécifique n’a été rapporté avec ENDOBULINE, la survenue de certains effets secondaires (voir paragraphe “Quels sont les effets indésirables éventuels”) dose-dépendants pourrait être favorisée : méningite, insuffisance rénale (anomalie de la fonction rénale), hyperviscosité sanguine.
Instructions en cas d'omission d'une ou de plusieurs doses
Si vous oubliez de prendre d'ENDOBULINE 50 mg/ml: ne prenez pas de dose double pour compenser la dose simple que vous avez oublié de prendre.
Sans objet.
4. QUELS SONT LES EFFETS INDÉSIRABLES ÉVENTUELS ?
Description des effets indésirables
Comme tous les médicaments, ENDOBULINE 50 mg/ml est susceptible d'avoir des effets indésirables.
Les effets secondaires liés à l'administration d'immunoglobuline normale sont plus fréquents chez les malades atteints de déficits immunitaires primitifs.
·Comme avec les autres immunoglobines humaines normales, des réactions de type frissons-hyperthermie (augmentation de la température du corps) parfois accompagnées de céphalées (maux de tête), de nausées (envies de vomir), de vomissements, de manifestations allergiques, d'élévation ou de chute de la pression artérielle, d'arthralgies (douleurs au niveau des articulations) et lombalgies (douleurs lombaires) modérées peuvent survenir occasionnellement.
intraveineuse rapide (voir paragraphe Mode d'administration) chez des patients agammaglobulinémiques avec déficit en IgA ou hypogammaglobulinémiques (avec défaut de production d'anticorps) qui n'ont jamais reçu ENDOBULINE ou dont le dernier traitement par ENDOBULINE remonte à plus de 8 semaines.
·Comme avec les autres préparations d'Immunoglobuline humaine normale, de rares cas de réactions cutanées surtout eczématiformes, régressives, de rares cas d'anémie hémolytique (diminution du ombre de globules rouges dans le sang par destruction) et/ou hémolyse (destruction des globules rouges) régressive et des cas d'augmentation de la créatininémie (mesure de fonctionnement du rein), et/ou d'insuffisance rénale aiguë (anomalie de la fonction rénale) ont été rapportés.
Si vous remarquez des effets indésirables non mentionnés dans cette notice, veuillez en informer votre médecin ou votre pharmacien.
5. COMMENT CONSERVER ENDOBULINE 50 mg/ml, poudre et solvant pour solution injectable?
Conditions de conservation et date de péremption
Tenir hors de la portée et de la vue des enfants.
A conserver au réfrigérateur (entre + 2 °C et + 8 °C) et à l'abri de la lumière.
Ne pas congeler.
Après reconstitution, la solution doit être utilisée immédiatement ou dans les 24 heures si elle est conservée à une température ne dépassant pas + de 25° C.
Ne pas utiliser après la date de péremption figurant sur l'étiquette
Si nécessaire, mises en garde contre certains signes visibles de détérioration
N'utilisez pas ENDOBULINE si vous constatez que la solution est trouble ou qu’elle contient un dépôt.
La dernière date à laquelle cette notice a été approuvée est le {date}